Содержание
- 0.1 Патогенез аутосомно-доминантного поликистоза почек (АДПКП)
- 0.2 Фенотип и развитие аутосомно-доминантного поликистоза почек (АДПКП)
- 0.3 Лечение аутосомно-доминантного поликистоза почек (АДПКП)
- 0.4 Риски наследования аутосомно-доминантного поликистоза почек
- 0.5 Патогенез аутосомно-доминантного поликистоза почек (АДПКП)
- 0.6 Фенотип и развитие аутосомно-доминантного поликистоза почек (АДПКП)
- 0.7 Лечение аутосомно-доминантного поликистоза почек (АДПКП)
- 0.8 Риски наследования аутосомно-доминантного поликистоза почек
- 1 Причины
- 2 Патофизиология
- 3 Симптомы и признаки
- 4 Диагностика
- 5 Прогноз
- 6 Лечение
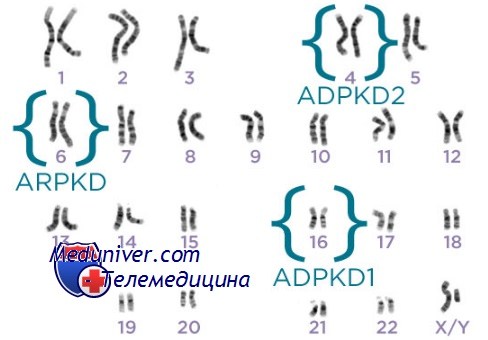
Этиология и встречаемость поликистоза почек. Аутосомно-доминантный поликистоз почек (АДПКП) (MIM №173900) — генетически разнородное заболевание. Приблизительно у 85% пациентов поликистоз I типа вызван мутациями в гене PKD1; большинство остальных имеют поликистоз II типа вследствие мутаций в гене PKD2. Несколько семей не показывают сцепления с этим локусами, что позволяет предполагать наличие, по крайней мере, еще одного дополнительного, пока неопознанного локуса.
Аутосомно-доминантный поликистоз почек (АДПКП) — одно из наиболее частых генетических заболеваний, имеет распространенность от 1 на 300 до 1 на 1000 во всех изученных этнических группах. В США заболевание составляет 8-10% терминальной почечной недостаточности.
Патогенез аутосомно-доминантного поликистоза почек (АДПКП)
Ген PKD1 кодирует полицистин-1, трансмембранный рецептороподобный белок неизвестной функции. Ген PKD2 кодирует полицистин-2, мембранный белок, гомологичный натриевому и кальциевому а1 каналам. Полицистин-1 и полицистин-2 взаимодействуют как часть гетеромногомерного комплекса.
Образование кист при аутосомно-доминантном поликистозе почек (АДПКП), как оказалось, соответствует «двухударному» механизму, наблюдающемуся при мутациях генов-супрессоров опухолевого роста и новообразованиях; т.е. для формирования кист должны утратить функцию оба аллеля гена PKD1 или PKD2. Механизм функциональной причины образования кисты при утрате функций полицистинов до конца не определен, но включает неправильное расположение белков на поверхности клетки, в норме ограниченное базолатеральной или эпителиальной поверхностью формирующихся тубулярных клеток почки.
Фенотип и развитие аутосомно-доминантного поликистоза почек (АДПКП)
Аутосомно-доминантный поликистоз почек (АДПКП) может обнаруживаться в любом возрасте, но симптомы или признаки чаще появляются на третьем-четвертом десятилетиях жизни. У пациентов появляются инфекции мочевых путей, гематурия, нарушения уродинамики (сгустки или камни почек), ноктурия, кровоизлияния в кисты или жалобы на боли в боку, вызванные увеличением размеров расширенных почек. Артериальная гипертензия бывает у 20-30% детей и почти у 75% взрослых с аутосомно-доминантным поликистозом почек. Гипертония — вторичный эффект внутрипочечной ишемии и активизации системы ренин-ангиотензин. Почти половина больных к 60 годам жизни имеет почечную недостаточность. Гипертония, повторные инфекции мочевых путей, мужской пол и возраст начала клинических проявлений — наиболее важные факторы, указывающие на раннее развитие почечной недостаточности. Приблизительно 43% пациентов с началом болезни вскоре после рождения умирают от почечной недостаточности в течение первого года жизни; у остальных к 30 годам жизни формируется терминальная почечная недостаточность, гипертония или и то, и другое вместе.
Аутосомно-доминантный поликистоз почек (АДПКП) демонстрирует как межсемейную, так и внутрисемейную изменчивость по возрасту начала и тяжести. Часть междусемейной изменчивости вторична относительно локусной гетерогенности, поскольку пациенты с поликистозом II типа имеют более мягкие проявления болезни, чем пациенты с I типом болезни. Внутрисемейная изменчивость, как оказалось, вызвана совместным влиянием среды и генетического фона, поскольку изменчивость более выражена между поколениями, чем среди сибсов.
Кроме кист почек, у пациентов с аутосомно-доминантным поликистозом почек образуются кисты в печени, поджелудочной железе, яичниках и селезенке, а также внутричерепные аневризмы, пролапс митрального клапана и дивертикулы толстой кишки. Кисты печени часто встречаются как при АДПКП-1, так и при АДПКП-2, тогда как кисты поджелудочной железы обычно наблюдают при АДПКП-1. Внутричерепные мешотчатые аневризмы выявляют у 5-10% больных АДПКП; тем не менее риск развития аневризм неодинаков для всех пациентов, поскольку они имеют семейное накопление. Больные аутосомно-доминантным поликистозом почек имеют повышенный риск недостаточности аортального и трехстворчатого клапанов, а пролапс митрального клапана выявляют у 25% больных. Дивертикулы толстого кишечника — наиболее частая внепочечная аномалия; при этом разрыв дивертикула при аутосомно-доминантном поликистозе почек более вероятен, чем при дивертикулах, наблюдаемых в общей популяции.
Особенности фенотипических проявлений аутосомно-доминантного поликистоза почек (АДПКП):
• Возраст начала: от детства до зрелости
• Прогрессирующая почечная недостаточность
• Почечные и печеночные кисты
• Внутричерепные мешотчатые аневризмы
• Пролапс митрального клапана
• Дивертикулы толстого кишечника
Лечение аутосомно-доминантного поликистоза почек (АДПКП)
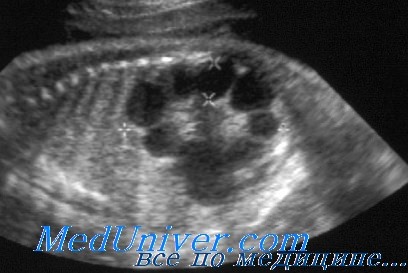
В основном аутосомно-доминантный поликистоз почек диагностируют по семейному анамнезу и УЗИ почек. Обнаружение почечных кист при УЗИ увеличивается с возрастом, так что 80-90% пациентов имеют обнаруживаемые кисты к 20 годам и почти 100% — к 30 годам жизни. Если это необходимо для пренатальной диагностики или идентификации родственника-донора почки, диагноз можно подтвердить анализом сцепления или прямым обнаружением мутации, или, в некоторых семьях, и тем, и другим.
Оказание помощи и лечение больных аутосомно-доминантным поликистозом почек нацелено на задержку развития почечной недостаточности и коррекцию симптомов. Гипертонию и инфекции мочевых путей следует энергично лечить, чтобы сохранить функцию почек. Боли, вызванные увеличением почек, уменьшаются при дренировании и склерозировании кист.
Риски наследования аутосомно-доминантного поликистоза почек
Приблизительно 90% пациентов имеют аутосомно-доминантный поликистоз почек в семейном анамнезе; только 10% аутосомно-доминантного поликистоза почек возникает вследствие новых мутаций в генах PKD1 или PKD2. Родители с аутосомно-доминантным поликистозом почек имеют 50% риск родить больного ребенка при каждой беременности. Если родители имели ребенка с внутриутробным началом болезни, риск родить другого сильно пораженного ребенка составляет приблизительно 25%. Тем не менее точно предсказать тяжесть проявлений болезни невозможно из-за варьирующей экспрессивности. Для семей, в которых известна мутация или возможен анализ сцепления, риск повторения может быть изменен при анализе ДНК плода.
Сибсы и родители пациентов с аутосомно-доминантным поликистозом почек также имеют повышенный риск болезни. Для обследования членов семьи рекомендуемый метод — ультрасонография почек.
Пример аутосомно-доминантного поликистоза почек (АДПКП). П.Д., 35-летний мужчина с пролапсом митрального клапана в анамнезе, поступает в местное отделение неотложной помощи с выраженными болями в боку и гематурией. Четыре месяца тому назад у него появились эпизодические боли в боку. УЗИ почек выявило нефролитиаз и множественные кисты почек, характерные для поликистоза почек. Данные его клинического осмотра в норме, за исключением систолического шума, соответствующего пролапсу митрального клапана, небольшой артериальной гипертензии и легкого повышения концентрации креатинина в сыворотке. Его отец и сестра умерли от прорыва внутричерепных аневризм, а сын умер в 1 год от поликистоза почек. После смерти сына врачи предлагали обследовать его и его жену на наличие поликистоза почек; однако родители решили не обследоваться из-за чувства вины и горя, вызванных смертью сына. Пациенту начато лечение камней в почках. Во время лечения нефролог сообщил больному, что у него аутосомно-доминантный поликистоз почек (АДПКП).
Этиология и встречаемость поликистоза почек. Аутосомно-доминантный поликистоз почек (АДПКП) (MIM №173900) — генетически разнородное заболевание. Приблизительно у 85% пациентов поликистоз I типа вызван мутациями в гене PKD1; большинство остальных имеют поликистоз II типа вследствие мутаций в гене PKD2. Несколько семей не показывают сцепления с этим локусами, что позволяет предполагать наличие, по крайней мере, еще одного дополнительного, пока неопознанного локуса.
Аутосомно-доминантный поликистоз почек (АДПКП) — одно из наиболее частых генетических заболеваний, имеет распространенность от 1 на 300 до 1 на 1000 во всех изученных этнических группах. В США заболевание составляет 8-10% терминальной почечной недостаточности.
Патогенез аутосомно-доминантного поликистоза почек (АДПКП)
Ген PKD1 кодирует полицистин-1, трансмембранный рецептороподобный белок неизвестной функции. Ген PKD2 кодирует полицистин-2, мембранный белок, гомологичный натриевому и кальциевому а1 каналам. Полицистин-1 и полицистин-2 взаимодействуют как часть гетеромногомерного комплекса.
Образование кист при аутосомно-доминантном поликистозе почек (АДПКП), как оказалось, соответствует «двухударному» механизму, наблюдающемуся при мутациях генов-супрессоров опухолевого роста и новообразованиях; т.е. для формирования кист должны утратить функцию оба аллеля гена PKD1 или PKD2. Механизм функциональной причины образования кисты при утрате функций полицистинов до конца не определен, но включает неправильное расположение белков на поверхности клетки, в норме ограниченное базолатеральной или эпителиальной поверхностью формирующихся тубулярных клеток почки.
Фенотип и развитие аутосомно-доминантного поликистоза почек (АДПКП)
Аутосомно-доминантный поликистоз почек (АДПКП) может обнаруживаться в любом возрасте, но симптомы или признаки чаще появляются на третьем-четвертом десятилетиях жизни. У пациентов появляются инфекции мочевых путей, гематурия, нарушения уродинамики (сгустки или камни почек), ноктурия, кровоизлияния в кисты или жалобы на боли в боку, вызванные увеличением размеров расширенных почек. Артериальная гипертензия бывает у 20-30% детей и почти у 75% взрослых с аутосомно-доминантным поликистозом почек. Гипертония — вторичный эффект внутрипочечной ишемии и активизации системы ренин-ангиотензин. Почти половина больных к 60 годам жизни имеет почечную недостаточность. Гипертония, повторные инфекции мочевых путей, мужской пол и возраст начала клинических проявлений — наиболее важные факторы, указывающие на раннее развитие почечной недостаточности. Приблизительно 43% пациентов с началом болезни вскоре после рождения умирают от почечной недостаточности в течение первого года жизни; у остальных к 30 годам жизни формируется терминальная почечная недостаточность, гипертония или и то, и другое вместе.
Аутосомно-доминантный поликистоз почек (АДПКП) демонстрирует как межсемейную, так и внутрисемейную изменчивость по возрасту начала и тяжести. Часть междусемейной изменчивости вторична относительно локусной гетерогенности, поскольку пациенты с поликистозом II типа имеют более мягкие проявления болезни, чем пациенты с I типом болезни. Внутрисемейная изменчивость, как оказалось, вызвана совместным влиянием среды и генетического фона, поскольку изменчивость более выражена между поколениями, чем среди сибсов.
Кроме кист почек, у пациентов с аутосомно-доминантным поликистозом почек образуются кисты в печени, поджелудочной железе, яичниках и селезенке, а также внутричерепные аневризмы, пролапс митрального клапана и дивертикулы толстой кишки. Кисты печени часто встречаются как при АДПКП-1, так и при АДПКП-2, тогда как кисты поджелудочной железы обычно наблюдают при АДПКП-1. Внутричерепные мешотчатые аневризмы выявляют у 5-10% больных АДПКП; тем не менее риск развития аневризм неодинаков для всех пациентов, поскольку они имеют семейное накопление. Больные аутосомно-доминантным поликистозом почек имеют повышенный риск недостаточности аортального и трехстворчатого клапанов, а пролапс митрального клапана выявляют у 25% больных. Дивертикулы толстого кишечника — наиболее частая внепочечная аномалия; при этом разрыв дивертикула при аутосомно-доминантном поликистозе почек более вероятен, чем при дивертикулах, наблюдаемых в общей популяции.
Особенности фенотипических проявлений аутосомно-доминантного поликистоза почек (АДПКП):
• Возраст начала: от детства до зрелости
• Прогрессирующая почечная недостаточность
• Почечные и печеночные кисты
• Внутричерепные мешотчатые аневризмы
• Пролапс митрального клапана
• Дивертикулы толстого кишечника
Лечение аутосомно-доминантного поликистоза почек (АДПКП)
В основном аутосомно-доминантный поликистоз почек диагностируют по семейному анамнезу и УЗИ почек. Обнаружение почечных кист при УЗИ увеличивается с возрастом, так что 80-90% пациентов имеют обнаруживаемые кисты к 20 годам и почти 100% — к 30 годам жизни. Если это необходимо для пренатальной диагностики или идентификации родственника-донора почки, диагноз можно подтвердить анализом сцепления или прямым обнаружением мутации, или, в некоторых семьях, и тем, и другим.
Оказание помощи и лечение больных аутосомно-доминантным поликистозом почек нацелено на задержку развития почечной недостаточности и коррекцию симптомов. Гипертонию и инфекции мочевых путей следует энергично лечить, чтобы сохранить функцию почек. Боли, вызванные увеличением почек, уменьшаются при дренировании и склерозировании кист.
Риски наследования аутосомно-доминантного поликистоза почек
Приблизительно 90% пациентов имеют аутосомно-доминантный поликистоз почек в семейном анамнезе; только 10% аутосомно-доминантного поликистоза почек возникает вследствие новых мутаций в генах PKD1 или PKD2. Родители с аутосомно-доминантным поликистозом почек имеют 50% риск родить больного ребенка при каждой беременности. Если родители имели ребенка с внутриутробным началом болезни, риск родить другого сильно пораженного ребенка составляет приблизительно 25%. Тем не менее точно предсказать тяжесть проявлений болезни невозможно из-за варьирующей экспрессивности. Для семей, в которых известна мутация или возможен анализ сцепления, риск повторения может быть изменен при анализе ДНК плода.
Сибсы и родители пациентов с аутосомно-доминантным поликистозом почек также имеют повышенный риск болезни. Для обследования членов семьи рекомендуемый метод — ультрасонография почек.
Пример аутосомно-доминантного поликистоза почек (АДПКП). П.Д., 35-летний мужчина с пролапсом митрального клапана в анамнезе, поступает в местное отделение неотложной помощи с выраженными болями в боку и гематурией. Четыре месяца тому назад у него появились эпизодические боли в боку. УЗИ почек выявило нефролитиаз и множественные кисты почек, характерные для поликистоза почек. Данные его клинического осмотра в норме, за исключением систолического шума, соответствующего пролапсу митрального клапана, небольшой артериальной гипертензии и легкого повышения концентрации креатинина в сыворотке. Его отец и сестра умерли от прорыва внутричерепных аневризм, а сын умер в 1 год от поликистоза почек. После смерти сына врачи предлагали обследовать его и его жену на наличие поликистоза почек; однако родители решили не обследоваться из-за чувства вины и горя, вызванных смертью сына. Пациенту начато лечение камней в почках. Во время лечения нефролог сообщил больному, что у него аутосомно-доминантный поликистоз почек (АДПКП).
Поликистозная болезнь почек (ПБП) – заболевание с формированием почечных кист, вызывающее постепенное увеличение почек.
Клинические проявления включают боли в боку и в области живота. Диагноз устанавливается с помощью КТ или УЗИ. Лечение симптоматическое до стадии почечной недостаточности.
Причины
Наследование ПБП может быть аутосомно-доминантным или рецессивным, спорадические случаи встречаются редко. Распространенность аутосомно-доминантной поликистозной болезни почек (АДПБП) составляет 1/1000, или 5% пациентов с терминальной стадией заболевания почек, требующей заместительной терапии. Клинические проявления редко наступают до достижения взрослого возраста, но наблюдается полная пенетрантность; все пациенты старше 80 лет имеют некоторые клинические проявления. Напротив, аутосомно-рецессивная ПБП встречается редко, ее распространенность 1/10000. Она часто вызывает почечную недостаточность в детстве.
В 86-96% случаев АДПБП вызвана мутациями в гене PKD1 на 16-й хромосоме, который кодирует белок полицистин 1.
Патофизиология
Полицистин 1 регулирует адгезию и дифференцировку эпителиальных клеток канальцев; полицистин 2 может работать как ионный канал с мутациями, вызывающими секрецию жидкости в кистах. Мутации этих белков могут изменять функцию почечных ресничек, что позволяет клеткам канальцев реагировать на скорость потока мочи. Ведущая гипотеза предполагает, что пролиферация клеток канальцев и дифференцировка связаны со скоростью потока мочи, и цилиарная дисфункция может,таким образом, приводить к кистозной трансформации.
На ранних стадиях заболевания происходит дилатация канальцев и медленное наполнение их гломерулярным фильтратом. В результате канальцы отделяются от функционирующих нефронов и наполняются жидкостью в результате ее секреции, а не фильтрации, формируя кисты. Может происходить кровоизлияние в кисты, вызывая гематурию. В результате неизвестных механизмов развивается сосудистый склероз и интерстициальный фиброз.
Внепочечные проявления встречаются часто:
- У большинства пациентов имеются кисты печени.
- У пациентов также часто встречаются панкреатические и кишечные кисты, дивертикулы толстой кишки, паховые грыжи.
- Пороки клапанов сердца могут быть обнаружены с помощью ЭХО-КГ у 25-30% пациентов, другие заболевания клапанов могут быть связаны с нарушениями обмена коллагена, и
- Аортальная регургитация вследствие расширения корня аорты в результате артериальных изменений стенок сосудов (включая аневризму аорты).
- Встречаются аневризмы коронарных артерий.
- Около 4% молодых и до 10% пожилых пациентов имеют мозговые аневризмы. Разрыв аневризмы случается в 65-75% случаев, обычно в возрасте до 50 лет.
Симптомы и признаки
Обычно АДПБП изначально протекает бессимптомно; у половины пациентов заболевание и в дальнейшем остается бессимптомным и не диагностированным. Большое количество пациентов, у которых развиваются клинические признаки заболевания, на момент их манифестации достигают возраста 20-30 лет. Клиническая картина включает боли в нижне-боковой области, в области живота и в нижней части спины, вызванные ростом кисты и симптомами инфекции. Если случается приступ острой боли, то он, как правило, вызван кровоизлиянием в кисту или прохождением камня; лихорадка часто сопровождает острый пиелонефрит. Пороки клапанов сердца редко проявляются клиническими симптомами, но иногда вызывают сердечную недостаточность и требуют замены клапана. Мозговые аневризмы до разрыва могут не проявлять себя, но могут являться причиной головных болей,тошноты, рвоты и дефицита черепно-мозговых нервов; эти клинические проявления свидетельствуют о необходимости срочного вмешательства.
Лабораторные признаки неспецифичны и включают гематурию и артериальную гипертензию (каждое проявление встречается в 40-50% случаев) и протеинурию (в 20%). Анемия встречается реже, чем при других типах хронической почечной недостаточности, в основном потому, что образование эритропротеина сохранено.
Диагностика
- УЗИ.
- Иногда КТ или МРТ или генетическое исследование.
Диагноз можно заподозрить у пациентов со следующими признаками:
- Семейный анамнез заболевания.
- Типичная клиническая картина.
- Случайно выявленные при визуализирующих методах исследования кисты.
Необходимо консультировать пациентов до назначения диагностических процедур, особенно при бессимптомном течении. Например, многие специалисты не рекомендуют обследование молодых пациентов с бессимптомным течением, потому что не существует эффективного лечения на этой стадии, а установление диагноза оказывает потенциальное негативное воздействие на получение страхового полиса и настроение пациента. Диагноз обычно устанавливается визуализирующими методами, демонстрируя распространенные кистозные изменения в почках и картинку «изъеденных молью» тканей, из-за кист, которые смещают функциональную ткань. Эти изменения развиваются с возрастом и редко обнаруживаются у молодых пациентов. Первым исследованием обычно проводится УЗИ. Если по результатам УЗИ нельзя сделать заключение, проводится КТ или МРТ (оба метода более чувствительные, особенно при использовании контрастирования).
В анализе мочи можно определить умеренную протеинурию и микроскопическую или макроскопическую гематурию. Макрогематурия может быть вызвана сместившимся камнем или кровоизлиянием из разорвавшейся кисты. Пиурия часто встречается и без бактериальной инфекции. Изначально, уровни азота мочевины крови и креатинина нормальные или только слегка повышены, особенно при наличии артериальной гипертензии. Иногда в общем анализе крови выявляется полицитемия.
Пациентам с симптомами мозговой аневризмы требуется КТ высокого разрешения или МР-ангиография. Однако не существует консенсуса по вопросу, должны ли пациенты с бессимптомным течением проходить скрининг на наличие мозговых аневризм. Рациональный подход предполагает проводить скрининг среди пациентов с АДПБП и семейным анамнезом геморрагических инсультов и мозговых аневризм.
Генетическое исследование для определения мутаций в гене PKD в настоящее время проводится только в следующих случаях:
- Пациентам с подозрением на ПБП и без известного семейного анамнеза.
- Пациентам с сомнительными результатами визуализирующих методов исследования.
- Наиболее молодым пациентам (например, возраст моложе 30 лет, для которых результаты визуализирующих методов исследования часто сомнительные).
Прогноз
К возрасту 75 лет 50-75% пациентов с АДПБП требуется заместительная терапия почечной недостаточности. Существуют следующие предпосылки к более быстрому прогрессированию почечной недостаточности:
- Более молодой возраст на момент установления диагноза.
- Мужской пол.
- Серповидно-клеточная аномалия эритроцитов.
- PKD1 генотип.
- Значительное или быстрое увеличение почек в размерах.
- Макрогематурия.
- Артериальная гипертензия.
АДПБП не повышает риск рака почки, но если у пациента с АДПБП развивается рак почки, он чаще бывает двусторонним. Рак почки редко является причиной смерти. Пациенты обычно умирают от сердечной патологии (иногда клапанной), диссеминированной инфекции или разрыва аневризмы.
Лечение
- Контроль факторов риска.
- Поддерживающие мероприятия.
Необходим строгий контроль АД, своевременное лечение ИМП. Чрескожная аспирация содержимого кист помогает бороться с сильными болями вследствие кровоизлияния или компрессии кист. Нефрэктомию выполняют для облегчения тяжелых симптомов в результате значительного увеличения почки (таких, как боль, гематурия) или рецидивирующих ИМП. АДПБП не рецидивирует в трансплантате. В процессе диализа у пациентов с АДПБП удается поддерживать более высокий уровень гемоглобина.