Содержание
Синдромом Поттера называют серьезный дефект развития плода. Характеристику начнем с определения этой аномалии.
Что такое Potter Syndrome?
Синдром Поттера – врожденный дефект развития плода, при котором у него полностью отсутствуют почки, что является следствием уменьшения объема околоплодных жидкостей.
Результатом маловодия является сдавливание плода в утробе матери. У детей также наблюдаются плохо развитые легкие, типичное сморщенное лицо и сдавленный череп, деформированные конечности.
В 1964 году Поттером был описан дефект развития, характеризующийся агенезией (отсутствием развития) обеих почек плода или аплазией (отсутствием какого-либо органа или части тела), сочетающейся с аномалиями лица. Данный синдром упоминается в медицинской литературе так же как синдром Поттера с агенезией 2 почек, почечно-лицевой синдром Гросс, лицевая почечная дисплазия.
Синдром Поттера у плода: причины
Этиология (происхождение) синдрома все еще не изучена до конца. В 50% случаев было выявлено, что первичная аномалия проявляется из-за отсутствия амниотической жидкости (плодных вод) у беременной женщины. Сдавливание плода маткой приводит к развитию аномалий почек, лица (сплющивание и уплощение), сердца, прямой кишки, гениталий, легких (гипоплазия), конечностей (косолапость).

Встречается синдром Поттера у грудничков, детей в возрасте нескольких месяцев, реже – у малышей старше года, причем чаще всего у мальчиков. Частота проявления дефекта: 1 на 50 тыс. родов.
Обследование родителей
Какими симптомами сопровождается синдром Поттера у плода? Большие белые почки – это то, что можно увидеть при ультразвуковом исследовании. Такая агенезия может быть доминантно наследованной (наследственность другого родителя не может ее подавить), т.е. не связанной с количеством околоплодных вод. У одного из родителей может быть недоразвитие или отсутствие одной из почек, что могло быть упущено при медицинских обследованиях ранее.
Агенезия почек наследуется доминантно, а значит, существует 50% вероятность рождения плода, у которого будет наблюдаться синдром Поттера.
Синдром Поттера с 75-процентной вероятностью может проявиться у малыша, если у его родителей обнаружены мутации в гене PKHD1. Они встречаются в среднем у 1 из 50 человек. Подобные мутации не приводят к скорому развитию дефекта у носителя, они могут десятилетиями передаваться из поколения в поколение.
Риск рождения малыша с синдромом Поттера может проявиться при наследовании мутаций от обоих родителей. Если плоду передадутся генетические изменения только отца или матери, то с 50-процентной вероятностью он родится здоровым. 25% малышей вообще не наследуют мутации PKHD1 от обоих родителей, имеющих таковые изменения на генном уровне.
Симптоматика синдрома
Синдром Поттера у плода диагностика выявляет по следующим проявлениям:
- чрезвычайно узкие щели век;
- характерная борозда под линией века;
- недоразвитие нижней челюсти (микрогнация);
- приплюснутый носик;
- аномально большое расстояние между парными органами (в частности глазами) – гипертелоризм;
- выпуклый эпикантус ("монгольская складка") – специальная складка у внутреннего уголка глаза, прикрывающая слезный бугорок;
- аномальной формы мягкие большие уши.
Возможность лечения
При рождении ребенка главный симптом – тяжелая дыхательная недостаточность, которая проявляется с первых минут самостоятельной жизни. Проведение искусственной вентиляции легких осложняется пневмотораксом (наличием пузырьков воздуха в плевральной области, которые затрудняют ритмичное движение легких).
К сожалению, подавляющая часть новорожденных, у которых диагностирован синдром Поттера, погибают через несколько часов после родов. При более легких его формах, касающихся дигестивного (пищеварительного) тракта – мочеполовая клоака (соединение анального и мочеполового протока в один), отсутствие перфорации ануса (правильного оформления) – применяется хирургическое вмешательство.
Выживший ребенок с синдромом Поттера
В 2013 году в СМИ появилась информация о новорожденной девочке Эбигейл Бутлер, дочери члена Палаты Представителей парламента Соединенных Штатов, республиканской конгрессвумен Хайме Эррер-Бутлер. Ребенок смог выжить с этим синдромом, препятствующим нормальному функционированию дыхательной системы.
На 5-м месяце беременности женщина узнала, что у девочки не выделяется моча из-за полного отсутствия почек. Причиной был дефицит околоплодных вод у беременной Хайме Эррер-Бутлер. Несмотря на заключение врачей, объявивших этот случай фатальным, женщина вместе с мужем решила сохранить беременность. Чтобы как-то компенсировать малое количество околоплодных жидкостей, в матку Хайме вводили специальный солевой раствор. Джон Хопкинс – доктор, проводивший эту терапию, не мог стопроцентно гарантировать супругам, что лечение приведет к удовлетворительному результату.
Но 15 июля 2013 года маленькая Эбигейл родилась живой. По воспоминаниям матери, девочка не кричала при рождении, но спустя время заплакала – легкие ребенка функционировали. После рождения она сразу же был переведена на диализ – систему, полностью заменяющую почечные функции, выводящую продукты обмена веществ из организма. Через год она заменяется донорскими почки.
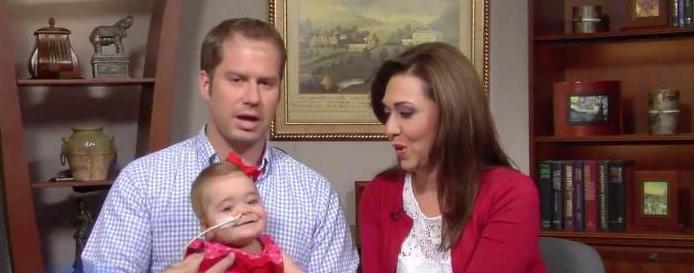
Шанс на рождение живого малыша с синдромом Поттера и его последующей успешной реабилитации в условиях современных реалий пока считается чудом. Поэтому медики настаивают на прерывание беременности при диагностировании у плода такого дефекта. Пример Эбигейл Бутлер не может не обнадеживать, но стоит понимать, что это исключительный случай.
Синонимы синдрома Поттера I. Почечно-лицевой синдром (Gross). Почечная агнозия (Potter).
Определение. Редкий, но типичный комплексный врожденный порок развития (почечно-лицевая дисплазия).
Авторы. Potter Edith L. — современный американский патолог, Чикаго. По-видимому, автором первого описания заболевания был Соеп (1884). Сообщение Potter было опубликовано в 1946 г.
Симптоматология синдрома Поттера I:
1. Характерная форма лица: гипертелоризм; выступающий лобный бугор; уплощение и расширение корня носа; большие, глубоко расположенные диспластические ушные раковины атипичной формы; микрогнатия; иногда старческое выражение лица новорожденного (так называемое лицо Potter). Иногда развивается гидроцефалия.
2. Пороки развития нижних конечностей: симбодия, анхиподия, сиреномелия, другие пороки развития стоп и т. д.
3. Пороки развития позвоночника: блоковидные и клиновидные позвонки; расщепление позвонков, особенно в верхней грудной, поясничной, а также в крестцовой областях.
4. Сочетание изменений, изложенных в п. 1 и 2, заставляет думать о возможности важнейших комплексных пороков развития: дисгенезии или агенезии почек или других пороков мочеобразующих и мочевыводящих органов с различной степенью выраженности и различными вариациями.
5. Пороки развития половых органов, особенно рудиментарное их развитие, атрезия, гермафродитизм, гипоспадия, uterus bicornis и др.
6. Гипоплазия или агенезия частей легких.
7. Могут встречаться и многочисленные другие пороки развития: атрезия гортани, пищевода, заднепроходного отверстия и прямой кишки; дисрафические расстройства развития головного мозга, четырехпалость и т. д.
8. Недостаточный вес при рождении.
9. Чаще болеют лица мужского пола.
10. Пороки развития часто бывают столь тяжелыми, что они в первые часы жизни приводят к смерти. Выживают обычно дети с ослабленными формами синдрома.
11. Часто обнаруживают также amnion nodosum и oligohydramnion.
Этиология и патогенез синдрома Поттера I. За исключением случаев заболевания близнецов, данных о семейном характере болезни нет. Отсутствуют также точные данные о роли наследственности. Пороки развития связаны, по-видимому, с поражением основной хорды в тератогенном детерминационном периоде, когда формируются самые ранние стадии развития.
Дифференциальный диагноз. Дисрафия. S. Greig (см.). S. Apert I (см.). S. Ullrich—Feichtiger (см.).

14. Харлоидное лицо – черты лица грубые, напоминающие гаргулий (увеличение языка и губ, зубы мелкие и далеко отстоят друг от друга). Сопровождается зачастую карликовым ростом, кифозом, деформациями конечностей (лопатообразные кисти рук), помутнением роговицы, гепатоспленомегалией, задержкой умственного развития, а так же больные могут отмечать ограничение подвижности суставов.
Если речь идет о синдроме Харлера (Гурлера), то он обуславливается недостатком альфа-L-идуронидазы , в результате чего в клетках происходит накопление патологических веществ, ведущие к серьезным поражениям хрящей и костей. Так же подобные изменения в чертах лица могут развиваться при мукополисахаридозах и муколипидозах.

15. Лицо Поттера – гипертелоризм глаз, высупающие эпикантические складки, ушные раковины низко посажены, подбородок недоразвит, нос уплощен. Подобные изменения характерны для двусторонней агенезии почек и других почечных аномалий.
16. Лицо при хронической почечной недостаточности – одутловатое лицо, напоминающее таковое при микседеме, но в отличие от последней, причинами возникновения являются безбелковые отеки, а не скопление коллагеновых волокон.

17. Микседематозное лицо – одутловатое, кожа шероховата, желтоватого оттенка. Желтизна обусловлена скоплением каротина. Брови в свой наружной трети имеют тенденцию к выпадению. Волосы становятся жесткими, глаза – мутными. Такое лицо характерно для микседемы (гипотиреоз).
18. Ладьевидное лицо – характеризуется впалой формой, которая напоминает блюдо: подбородок и лоб выступают, а нос и верхняя челюсть сплющены.
19. Лицо при симптоме Бэттла – кровоподтек под сосцевидным отростком, а иногда и за ним. Возникает в результате классического перелома основания черепа с кровоизлиянием в среднюю черепную ямку. Данный кровоподтек может появиться не сразу, а через 3-12 дней после травмы либо на стороне перелома, либо на противоположной. Чувствительность данного симптома не слишком велика: всего 2-8%, но прогностическое значение составляет 100%. 
20. Лицо при симптоме “очков” – кровоподтеки вокруг глазных орбит, возникающие при травме глаза, переломе черепа и внутричерепном кровотечении. Разновидность данного симптома может встречаться при амилоидозе в результате повышенной ломкости капилляров, которая наблюдается при данном заболевании. Проба Вальсальвы, при которой повышается центральное венозное давление,или обычная ректоскопия, которая приводит к этой пробе, могут служить провоцирующими факторами.
21. Лицо курильщика – черты грубые, кожа морщинистая, сероватая, атрофичная.

22. Склеродермическое лицо – нос на таком лице заострен, кожа тонкая и туго натянута вплоть до разглаживания морщин. Часто можно увидеть гиперпигментацию, сочетающуюся с участками витилиго и некоторым количеством телеангиэктозий. Характерен так же “кисетный рот”, при котором больные не могут широко открыть рот.
23. Лицо Хатчинсона (Гетчинсона) – веки на таком типе лица опущены (птоз), глазные яблоки неподвижны в результате офтальмоплегии. Больной компенсаторно запрокидывает голову назад.

24. Лицо при системной красной волчанке – характерны классические высыпания в области щек, переходящих на спинку носа в виде крыльев бабочки.
25. Миастеническое лицо – лицо, характерными чертами которого является птоз и опущенные уголки рта. В результате слабости лицевых мышц, на лице отмечается гипомимия и выражение аппатии. Миастения – заболевание, причина которого кроется в аутоиммунных расстройствах. Прогрессирующая слабость и утомляемость затрагивает скелетные мышцы. 
26. Миопатическое лицо – или иначе, лицо сфинкса. Очень похоже на лицо при миастении. Мышечная слабость провоцирует опущение век, общую релаксацию мышц лица и выпячивание губ. 
27. Тетаническое лицо – характеризуется сардонической улыбкой. При этом рот больного открыт, губы поперечно растянуты, а само лицо напоминает гримасу Джокера. Такое лицо можно увидеть при столбняке. Голова больного запрокинута, тело изогнуто дугой – вся мускулатура находится в сильном напряжении. При этом больной находится в сознании.
28. Лицо Паркинсона – можно наблюдать у пациентов с одноименной болезнью. Лицо амимично вплоть до маскообразности, застывшее и апатичное.

29. Лицо Грейвса – экзофтальм и неполное смыкание век, лицо встревоженное. Встречается при болезни Грейвса или базедова болезнь, при которой происходит выработка антител к рецептору ТТГ, с развитием тиреотоксикоза.

30. Акромегалическое лицо – для него характерно утолщение лицевых костей, нижняя челюсть и надбровные дуги выступают, нос и губы увеличены в размерах. Так же для акромегалии, нейроэндокринного заболевания с поражением гипофиза или/и гипоталамуса, характерно прогрессирующее увеличение головы, кистей, лица и стоп.

31. Кушингоидное лицо – лунообразное, круглое, немного отечное и маслянистое. На таком лице могут появляться угри, а так же аллопеция, сочетающиеся с избыточным оволосением лица. Для таких больных характерны жировые подушечки на щеках, горб “бизона”, стрии и ожирение по центральному типу.